Did you get a sequence electropherogram that does not look right?
Did your reaction fail?
Are you thinking of submitting a new sample and want to know how to prepare it?
Automated Sanger sequencing remains the gold standard for single target DNA sequencing. Typically a robust technique, sequencing reactions can and do fail from time to time. Luckily, there are a few simple steps one can take in the preparation of a sample for sequencing to minimize failed or low-quality sequencing results. If these steps have been taken and a sequencing reaction still fails, there are specific ways to interpret your electropherogram and identify the cause of the failure. For the vast majority of sequencing reactions, simply repeating the same reaction with the same template and primer mixture will not resolve a failed sequence. We have prepared a guide to help you assess your sequencing data and to identify potential causes of a failed reaction.
-
Best Practices for Sample Preparation
- PCR product
- Do you have a single product for your PCR?
- why: multiple templates will give a mixed template sequencing reaction, low quality/quantity template may cause short sequencing reactions or failed sequencing
- recommendation: assess by gel electrophoresis for each template submitted for sequencing
- Have you ensured your negative control PCR reaction (no DNA PCR) is blank?
- why: multiple templates will give a mixed template sequencing reaction (if your negative has a band the same size as your target, you may have a mixed template in your target band)
- recommendation: assess by gel electrophoresis for each set of PCR reactions that provide template submitted for sequencing
- Is your PCR cleaned of all PCR reaction reagents (primers, enzyme, buffers etc)?
- why: both PCR primers will work in your sequencing reaction and produce a mixed template sequence, buffers will impact performance of sequencing enzyme mix
- recommendation: post—PCR cleanup with a cleanup kit such as Qiaquick (Qiagen)
- Quantification of concentration: We often see a few problems here that cane be easily overcome. One is that quantifying a PCR sample before purification will lead to incorrect QA of your sample. The template and primer DNA in the PCR tube will give you a spec reading regardless of whether you have a product in the tube. Another common problem is that when using a Nanodrop, if the range of A260 is not between 0.1 and 0.8, the reading will be inaccurate (sometimes by orders of magnitude). If your A260 is above 0.8, you must dilute your sample and re-spec. See image here:
- Do you have a single product for your PCR?
- Plasmid DNA
- Did your plasmid extraction work efficiently?
- why: no plasmid DNA; you may get a spec reading that indicates you have DNA in your sample, but it may not be plasmid but other contaminants, such as bacterial DNA; multiple plasmid templates will give a mixed template sequencing reaction; low quality/quantity template may cause short sequencing reactions or failed sequencing
- recommendation: assess by gel electrophoresis for each template submitted for sequencing
- Quantification of concentration: We often see a few problems here that cane be easily overcome. One is that quantifying a dirty plasmid prep will lead to incorrect QA of your sample. Dirty plasmid preparations often contain bacterial genomic DNA or RNA and you will get a viable spec reading regardless of whether you have a plasmid in the tube. Another common problem is that when using a Nanodrop, if the range of A260 is not between 0.1 and 0.8, the reading will be inaccurate (sometimes by orders of magnitude). If your A260 is above 0.8, you must dilute your sample and re-spec. See image below:
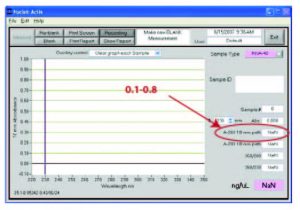
- Did your plasmid extraction work efficiently?
- PCR product
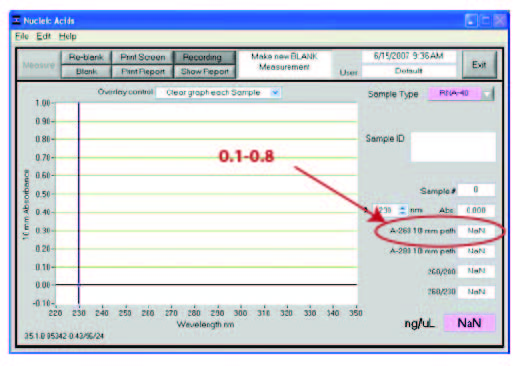
-
How to identify the cause of a sequencing failure
Helpful guides for identifying potential causes of sequencing failure from simply looking at the chromatogram can be found here:
Nucleics DNA Troubleshooting Guide
6 Tips for Analyzing and Troubleshooting DNA Sequencing Results